
This is a completely rewritten version of a biology paper titled, “A Model for the Evolution of the Mammalian T-cell Receptor α/δ and μ Loci Based on Evidence from the Duckbill Platypus.” It is meant to be a demonstration and a proof of concept for JAWWS, my idea of a science journal that focusses on readability.
You can read the original version on the Molecular Biology and Evolution journal’s website, or here on this blog, with annotations by me.
Why did I choose to rewrite this paper? I wish I had a more principled answer, but the truth is that I simply went to ResearchHub, a website where scientists share papers between themselves and upvote the most interesting one, went to the Evolutionary Biology section (because that used to be my field), and picked the first paper that was open-access and seemed fit for my purposes. In other words, a paper that seemed like it could be improved a lot because it seemed more difficult to understand than was warranted.
Only later did I realize it was a paper from 2012, so not that recent. I don’t think that matters too much for now since it’s just a proof of concept. Nor does the topic, i.e. molecular evolution in the vertebrate immune system. The actual journal will need to pick papers in a more principled way, of course.
What did my rewrite entail? The best way to know is to read (not necessarily closely) the original and rewritten versions. But here’s a sample of my “interventions”:
- I put almost all citations in collapsible footnotes.
- I cut up most long paragraphs, including the abstract.
- I added many context sentences, including at the beginning of sections, to give a better sense of why we’re reading this. One example is the first sentence of the introduction: “How did the immune system of jawed vertebrates evolve?”
- I reworked some of the paper’s structure. One major change: I put the major contribution of the study, that is, the new evolutionary model, in its own section after the introduction. This way, it is not buried deep in the discussion; readers can start with it and dig into the rest only if they want more details. I also reordered the methods so that they would match the ordering in the Results.
- I added several subheadings to the sections that didn’t already have subsections (Introduction, and Results and Discussion)
- I tried my best to avoid abbreviations. One difficulty is that some of them are probably very recognizable by people who know immunology, and not by me. So I left some in, while trying to make sure they don’t hamper readability. The major example is “TCR”, which means T-cell receptor and was used a lot. It’s still used in my version, but far less often.
- I removed some jargon words. E.g. “proximal” became “closest”.
- I formatted some information in point form, such as the T-cell lineages or the protocols in the Methods.
- I added some text formatting to guide the reader. For instance, bold font for groups of animals in the introduction, and color in the text to match colored elements in figures.
- I changed one figure by reorganizing its parts to make it clearer (fig. 2, which used to be fig. 5). A lot of additional clarity could potentially be gained from editing the figures, but that’s a lot of work, so I didn’t press it further.
- I fixed a number of typos and grammatical errors. Mistakes like this are not a big problem, but there were enough that I assume little editing work was done on this paper.
On footnotes: I’m using two different kinds of collapsible footnotes. Those in the usual style of the blog, like this one,1Here you would usually read a citation in short form such as “Rast et al. 1997.” Head to the original paper webpage to see the reference list in full. contain the citations included in the original paper. Footnotes with brackets like this[1]This is an example comment. are for comments on the rewriting process and are also shown at the bottom of the paper. I suggest you don’t click on the former, unless you want to see a reference, and hover onto the latter to read my comments.
Overall, my rewrite increased the length of the abstract from 267 to 286 words, and of the rest of the paper from about 6000 to about 6400 words. I consider this acceptable.
I will publish some more thoughts on the rewriting process later.
Abstract
Goal: This study presents a new model for the evolution of part of the vertebrate immune system: the genes encoding the T-cell receptor (TCR) δ chains.
Background: T lymphocytes have to recognize specific antigen for the adaptive immune response to work in vertebrates. They perform this using a somatically diversified T-cell receptor. All jawed vertebrates use four T-cell receptor chains called α, β, γ, and δ, but some lineages have nonconventional receptor chains: monotremes and marsupials encode a fifth one, called TCRµ. Its function is unknown, but it is somatically diversified like the conventional chains. Its origins are also unclear. It appears to be distantly related to the TCRδ chain, for which recent evidence from birds and frogs has provided new information that was not available from humans or other placental (eutherian) mammals.
Experiment: We analyzed the genes encoding the δ chains in the platypus. This revealed the presence of a highly divergent variable (V) gene, indistinguishable from immunoglobulin heavy chain V genes (VH), and related to V genes used in the µ chain. This gene is expressed as part of TCRδ repertoire, so it is designated VHδ.
Conclusions: The VHδ gene is similar to what has been found in frogs and birds, but it is the first time such a gene has been found in a mammal. This provides a critical link in reconstructing the evolutionary history of TCRµ. The current structure of the δ and µ genes in tetrapods suggests ancient and possibly recurring translocations of gene segments between the δ and immunoglobulin heavy genes, as well as translocations of δ genes out of the TCRα/δ locus early in mammals, creating the TCRµ locus. We present a detailed model of this evolutionary history.[2]Major changes to the abstract: I split it in four paragraphs with section titles (this is common in some journals; it should be common in most journals). I also added a section at the beginning to … Continue reading
Introduction
How did the immune system of jawed vertebrates evolve? In this study, we use genomic evidence from the platypus to propose a model for the evolution of a specific component of the vertebrate immune system: the receptors on the surface of T lymphocytes.
As a reminder, T lymphocytes (or T cells) are white blood cells that play a critical role in the adaptive immune system. They can be classified into two main lineages based on the receptor they use:2Rast et al. 1997; reviewed in Davis and Chein 2008
- αβT cell lineage: The receptor is composed of a heterodimer of α and β chains. Most circulating human T cells are αβT cells, including familiar subsets such as CD4+ helper T cells and regulatory T cells, CD8+ cytotoxic T cells, and natural killer T (NKT) cells.
- γδT cell lineage: The receptor is composed of γ and δ chains. The function of these cells is less well defined. They have been associated with a broad range of immune responses including tumor surveillance, innate responses to pathogens and stress, and wound healing.3Hayday 2009 γδT cells are found primarily in epithelial tissues and form a lower percentage of circulating lymphocytes in some species.
αβ and γδ T cells also differ in the way they interact with antigen. The receptors of αβT cells are “restricted” relative to the major histocompatibility complex (MHC), meaning that that they bind antigenic epitopes, such as peptide fragments, bound to, or “presented” by, molecules encoded in the MHC. In contrast, γδT receptors have been found to bind antigens directly in the absence of MHC, as well as self-ligands that are often MHC-related molecules.4Sciammas et al. 1994; Hayday 2009
All gnathostomes (jawed vertebrates) have αβ and γδ T cells. As we will see below, marsupial and monotreme mammals have an additional type of T-cell receptor, denoted with the letter µ. The platypus, a monotreme, further has a non-conventional receptor with δ chains, which is also present in birds and amphibians.
Before presenting our evolutionary model, let’s review these types of T-cell receptors and their structure.
Structure and Genes of Conventional T-Cell Receptors
The chains of conventional T-cell receptors are composed of two extracellular domains, both members of the immunoglobulin domain superfamily of cell surface proteins (fig. 1):5reviewed in Davis and Chein 2008
- The closest domain to the cellular membrane is called C for constant.[3]Since this abbreviation comes up a lot, I put it first, with its meaning in parentheses. The C domain is largely invariant among T-cell clones expressing the same class of the receptor chain.
- The domain farthest from the cellular membrane is called V for variable. It is the region that contacts antigen and MHC. Similar to antibodies, the individual clonal diversity in the V domain is generated by somatic DNA recombination.6Tonegawa 1983 [4]I didn’t change much in the figure’s caption, but it seemed pretty trivial to add color to the text to facilitate looking up what the colors mean.
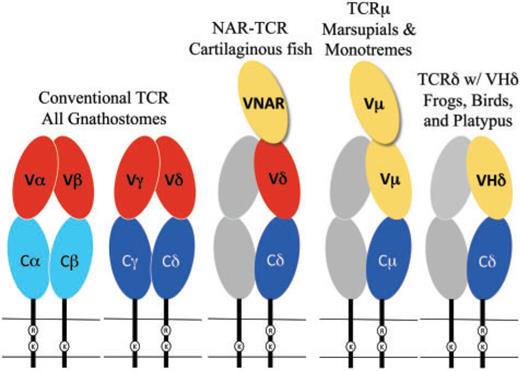
While C domains are usually encoded by a single, intact exon, V domains are assembled somatically from germ-line segments in developing T cells. These segments are genes called V (again for variable), D (for diversity), and J (for joining). The assembly process depends on the enzymes encoded by two genes, the recombination activating genes (RAG)-1 and RAG-2.7Yancopoulos et al. 1986; Schatz et al. 1989
The various T-cell receptor chains differ in how their V domains are assembled. β and δ chains are assembled from all three types of gene segments, whereas α and γ chains use only V and J. The different combinations of two or three segments, selected from a large repertoire of germ-line gene segments, along with variation at the junctions due to the addition and deletion of nucleotides during recombination, contribute to a vast diversity of T-cell receptors. It is this diversity that creates the individual antigen specificity of T-cell clones.[5]This is an example of two sentences taken verbatim from the original paper. Not all of it was poorly written!
These genes are highly conserved among species in both their genomic sequence and their organization.8Rast et al. 1997; Parra et al. 2008, 2012; Chen et al. 2009 In all tetrapods examined, the β and γ chains are each encoded at multiple separate loci, whereas the genes encoding the α and δ chains are nested at a single locus, called the TCRα/δ locus.9Chien et al. 1987; Satyanarayana et al. 1988; reviewed in Davis and Chein 2008 The V domains of α and δ chains can use a common pool of V gene segments, but distinct D, J, and C genes.
The recombination of V, J and optionally D genes, referred to as V(D)J recombination, and mediated by RAG, is also known to generate the diversity of antibodies produced by another type of lymphocyte, the B cells.[6]It took me forever to rewrite this part. The original sentence was, “Diversity in antibodies produced by B cells is also generated by RAG-mediated V(D)J recombination and the TCR and Ig genes … Continue reading10Flajnik and Kasahara 2010; Litman et al. 2010
Non-Conventional Receptors Across Vertebrates
VHδ genes have been found in both amphibians and birds (see the rightmost part of fig. 1).[8]Why not indicate the part of the figure that is relevant? Whenever you can, provide reader guidance! In the frog Xenopus tropicalis, as well as in a passerine bird, the zebra finch Taeniopygia guttata, the VHδ genes coexist with the conventional Vα and Vδ genes at the TCRα/δ locus.13Parra et al. 2010, 2012
In galliform birds, such as the chicken Gallus gallus, they are instead located at a second TCRδ locus that is unlinked to the conventional TCRα/δ.14Parra et al. 2012 VHδ are the only type of V gene segment present at the second locus and, although closely related to antibody VH genes, the VHδ appear to be used exclusively in δ chains. This is true as well for frogs where the TCRα/δ and IgH (immunoglobulin heavy chain) loci are tightly linked.15Parra et al. 2010
In mammals, a TCRα/δ locus has been characterized in several eutherian species and at least one marsupial, the opossum Monodelphis domestica. VHδ genes have not been found in mammals to date.16Satyanarayana et al.1988; Wang et al. 1994; Parra et al. 2008 However, marsupials do have an additional locus, unlinked to TCRα/δ, that uses antibody-related V genes. This fifth chain is called µ, and the receptor that uses it is referred to as TCRµ. The µ chain is related to the δ chain, but it diverges from it in both sequence and structure.17Parra et al. 2007, 2008 It has also been found in a monotreme, the platypus.[9]The authors like to use “duckbill platypus,” but there’s only one species of platypus, so I took that word out. The platypus and marsupial TCRµ genes are clearly orthologous, which is consistent with the idea that the µ chain is ancient in mammals, but has been lost in the eutherians.18Parra et al. 2008; Wang et al. 2011
TCRµ chains use their own unique set of V genes, called Vµ.19Parra et al. 2007; Wang et al. 2011 So far, no evidence has been found of V(D)J recombination between Vµ genes and genes from other immunoglobulin or T-cell receptor loci.[10]Another horrible sentence from the original, recorded for posterity: “Trans-locus V(D)J recombination of V genes from other Ig and TCR loci with TCRµ genes has not been found.” That … Continue reading Neither have TCRµ homologues been found in non-mammals.20Parra et al. 2008
The structure of TCRµ chains is atypical. They contain three, rather than two, extra-cellular domains from the immunoglobulin superfamily;[11]The abbreviation IgSF was used in the paper, with no explanation. I assume the people who would read this paper tend to know what that means, but still. this is due to an extra N-terminal V domain (see fig. 1).21Parra et al. 2007; Wang et al. 2011 Both V domains are encoded by a unique set of Vµ genes and are more related to immunoglobulin heavy chain V than to conventional T-cell receptor V domains. The N-terminal one is diverse and encoded by genes that undergo somatic V(D)J recombination, while the second V domain (referred to as “supporting”) has little or no diversity.
The supporting V domain differs between marsupials and monotremes. In marsupials, it is encoded by a germ-line joined, or pre-assembled, V exon that is invariant.22Parra et al. 2007 In the platypus, it is encoded by gene segments requiring somatic DNA recombination, but with limited diversity due in part to the lack of D segments.23Wang et al. 2011
Sharks and other cartilaginous fish also have a T-cell receptor chain that is structurally similar to TCRµ (see middle part of fig. 1).24Criscitiello et al. 2006; Flajnik et al. 2011 The resulting receptor is called NAR-TCR. Like the receptor of marsupials and monotremes, it contains three extracellular domains, but its N-terminal V domain is related to chains used by IgNAR (immunoglobulin new antigen receptor) antibodies, a type of antibody found only in sharks.25Greenberg et al. 1995 In both the TCRµ of marsupials and monotremes and the NAR-TCR of cartilaginous fishes, the current working model is that the N-terminal V domain is unpaired and acts as a single, antigen binding domain. This would be analogous to the V domains of light-chainless antibodies found in sharks and camelids.26Flajnik et al. 2011; Wang et al. 2011
How did the µ chain arise? Phylogenetic analyses support an origin after the avian–mammalian split.27Parra et al. 2007; Wang et al. 2011 Previously, we hypothesized that it originated as a recombination between ancestral immunoglobulin heavy and TCRδ-like loci,28Parra et al. 2008 but this hypothesis is problematic for several reasons. One challenge is the apparent genomic stability and ancient conserved synteny (order of genes on the chromosome) in the region surrounding the TCRα/δ locus; this region has appeared to remain stable over at least the past 350 million years of tetrapod evolution.29Parra et al. 2008, 2010
As a result, we need a new model for the evolution of TCRµ and the TCRα/δ locus. Here we present the best current model, supported by an analysis of the platypus genome—the first to examine a monotreme TCRα/δ locus in detail—as described in the methods and results sections below.
The Model
Our model can be summarized in six stages (fig. 2).[12]Major change from me here. This section was moved here from the discussion, because it is the core and most interesting part of the paper. It is now its own first-level section alongside … Continue reading

-
Duplication of the Dδ–Jδ–Cδ cluster. This occurred early in the evolution of tetrapods, or earlier. The duplication resulted in two copies of the C gene of the δ chain, each with its own set of D and J segments.
-
Insertion of VH. Recall that VH refers to the variable chain of immunoglobulin heavy (IgH). One or more genes were translocated from the IgH locus and inserted into the TCRα/δ locus, most likely to a location between the existing Vα/δ genes and the 5′-proximal Dδ–Jδ–Cδ cluster. This is the configuration found today in the zebra finch genome.30Parra et al. 2012
-
Inversion of the VHδ–Dδ–Jδ–Cδ cluster in amphibians. This cluster of genes was translocated and inverted, and the number of VHδ genes increased. The frog X. tropicalis currently has the greatest number of VHδ genes, where they make up the majority of V genes available in the germ-line for T-cell receptor δ chains.31Parra et al. 2010
-
Translocation of the VHδ–Dδ–Jδ–Cδ cluster to another site in galliforms. In chickens and turkeys, the same cluster that was inverted in amphibians instead moved out of the TCRα/δ locus and is now found on another chromosome. There are no Vα or Vδ genes at this second TCRδ locus in chickens, and only a single Cδ gene remains at the conventional TCRα/δ locus.32Parra et al. 2012
-
Translocation of the VHδ–Dδ–Jδ–Cδ cluster to another site (TCRµ) in mammals. A similar process to step D in galliforms happened in a common ancestor of mammals, giving rise to TCRµ. Internal duplications of the VH, D, and J genes gave rise to the current [(V–D–J) − (V–D–J) − C] organization that can encode chains with double V domains.33Parra et al. 2007, Wang et al. 2011
-
Further changes in the three mammalian lineages.
-
In the platypus, the second V–D–J cluster, which encodes the supporting (non-terminal) V chain, lost its D segments and generates V domains with short complementarity-determining region-3 (CDR3) encoded by direct V to J recombination.34Wang et al. 2011
-
Meanwhile, in therians (marsupials and placentals), the VHδ gene disappeared from the TCRα/δ locus (not shown in fig. 2).35Parra et al. 2008
-
Then, in placentals, the TCRµ locus was also lost.36Parra et al. 2008
-
The marsupials kept TCRµ, but the second set of V and J segments (which encode the supporting V domain) was replaced with a germ-line joined V gene (fused yellow–green segment in fig. 2), probably due to germ-line V(D)J recombination and retro-transposition.37Parra et al. 2007, 2008
-
In both monotremes and marsupials, the whole cluster from VH to C appears to have undergone additional tandem duplication as it exists in multiple copies in the opossum and probably in the platypus.38Parra et al. 2007, 2008; Wang et al. 2011
-
The rest of the paper explains the analyses that gave us with the evidence to build this model. Additional discussion of the model is provided in the last section.
Materials and Methods
There are three parts to the analyses and experiments that allowed us to gather evidence and build our evolutionary model. First, find the TCRα/δ locus in platypus genome data. Second, perform phylogenetic analyses with the relevant genes. Third, confirm from a live specimen that the platypus expresses VHδ.[13]This new paragraph is important! It gives context to the experiments below and it guides the reader for the entire section. Also notice this is a case of an enumeration without point form. I like … Continue reading
1) Identification and Annotation of the Platypus TCRα/δ Locus
We analyzed the genome of the platypus, Ornithorhynchus anatinus, using the assembly version 5.0.1 (http://www.ncbi.nlm.nih.gov/genome/guide/platypus/). We used two genome alignment tools: whole-genome BLAST from NCBI (www.ncbi.nlm.nih.gov/) and BLAST/BLAT from Ensembl (www.ensembl.org).
We located the V and J gene segments by looking for similarity with the corresponding segments of other species, and by identifying flanking conserved recombination signal sequences. (RSS). We annotated V segments in the 5′ to 3′ direction as either Vα or Vδ, followed by the family number and the gene segment number if there were more than one in the family. For example, Vα15.7 is the seventh Vα gene in family 15.
As for the D segments, we identified them from cDNA clones using VHδ, using complementarity-determining region-3 (CDR3) sequences that represent the V-D-J junctions.
We labeled the platypus T-cell receptor gene segments according to the IMGT nomenclature (http://www.imgt.org/). We provide the location for the TCRα/δ genes of the platypus genome version 5.0.1 in supplementary table S1, available online.
2) Phylogenetic Analyses
We used BioEdit39Hall 1999 as well as the accessory application ClustalX40Thompson et al. 1997 to align the nucleotide sequences of the V genes regions, from the framework region FR1 to FR3, including the complementarity-determining regions CDR1 and CDR2. We established the codon position of the alignments using amino acid sequences.41Hall 1999 When necessary, we corrected the alignments through visual inspection. We then analyzed them with MEGA Software.42Kumar et al. 2004
We generated phylogenetic trees using two methods: Neighbor Joining (NJ) with uncorrected nucleotide differences (p-distance), and Minimum Evolution distances.
We evaluated support for the generated trees using bootstrap values from 1000 replicates. Supplementary table S2 contains the GenBank accession numbers for the sequences used in tree construction.[14]In the original paper, this section comes after the Confirmation of Expression section below, but in the results section, the phylogenetic results are discussed first. I don’t know if there was … Continue reading
3) Confirmation of Expression of Platypus VHδ
As described with more detail in the Results and Discussion section below, the annotation step allowed us to find an atypical VHδ gene in the platypus genome. To confirm that it was not an artifact of the genome assembly process, we looked at the expression of this gene in a live specimen, a male platypus from the Upper Barnard River in New South Wales, Australia. The platypus was collected under the same permits as in Warren et al. 2008.
We performed reverse transcription PCR (RT-PCR) on the RNA from the spleen of this New South Wales specimen. As a second point of comparison, we also used a previously described platypus spleen cDNA library that was constructed from RNA extracted from a Tasmanian animal.43Vernersson et al. 2002 The protocols and products used at every step are as follows:
- cDNA synthesis: Invitrogen Superscript III-first strand synthesis kit, using the manufacturer’s recommended protocol44Invitrogen, Carlsbad, CA, USA
- PCR amplification: we used the QIAGEN HotStar HiFidelity Polymerase Kit45BD Biosciences, CLONTECH Laboratories, Palo Alto, CA, USA in total volume of 20 µl containing:
- 1× Hotstar Hifi PCR Buffer (containing 0.3 mM dNTPs)
- 1µM of primers: we identified these from the platypus genome assembly step.46Warren et al. 2008 We targeted T-cell receptor δ transcripts with two primers, one for VHδ and one for Cδ:
-
- 5′-GTACCGCCAACCACCAGGGAAAG-3′ for VHδ
- 5′-CAGTTCACTGCTCCATCGCTTTCA-3′ for Cδ
-
- 1.25U Hotstar Hifidelity DNA polymerase
- PCR product cloning: TopoTA cloning® kit 47Invitrogen
- Sequencing: BigDye terminator cycle sequencing kit version 348Applied Biosystems, Foster City, CA, USA according to the manufacturer recommendations.
- Analysis of sequencing reactions: ABI Prism 3100 DNA automated sequences.49PerkinElmer Life and Analytical Sciences, Wellesley, MA, USA
- Chromatogram analysis: Sequencher 4.9 software50Gene Codes Corporation, Ann Arbor, MI, USA
We archived the sequence on GenBank under the accession numbers JQ664690–JQ664710.
Results and Discussion
Results of the TCRα/δ Locus Identification in the Platypus
Here are the results of our analysis of the platypus genome from part 1 of the Materials and Methods section, which allowed us to identify the TCRα/δ locus and annotate its V, D, J and C gene segments, as well as the exons. Refer to fig. 3 below for the annotation map.

Most of the locus is present on a single scaffold. The remainder is on a shorter contig. On either sides of the locus, we find the genes SALL2, DAD1, and several olfactory receptor genes (OR). All of these genes share conserved synteny with the TCRα/δ locus in amphibians, birds, and mammals.51Parra et al. 2008, 2010, 2012
The platypus locus has many typical features common to TCRα/δ loci in other tetrapods.52Satyanarayana et al. 1988; Wang et al. 1994; Parra et al. 2008, 2010, 2012 Two C region genes are present: a Cα (light blue in fig. 3) at the 3′ end of the locus, and a Cδ (dark blue) oriented 5′ of the Jα genes. These Jα genes occur in a large number (32) of fragments (in green) located between Cδ and Cα. A large array of Jα genes like this is believed to facilitate secondary Vα to Jα rearrangements in developing αβT cells if the primary rearrangements are nonproductive or need replacement.53Hawwari and Krangel 2007 Primary TCRα V–J rearrangements generally use Jα segments towards the 5′-end of the array and can progressively use downstream Jα in subsequent rearrangements. There is also a single Vδ gene (the last red segment in fig. 3) in reverse transcriptional orientation between the platypus Cδ gene and the Jα array that is conserved in mammalian TCRα/δ both in location and orientation.54Parra et al. 2008
There are 99 conventional T-cell receptor V gene segments in the platypus TCRα/δ locus (red in fig. 3). The vast majority, 89, share nucleotide identity with Vα in other species; the other 10 share identity with Vδ genes. The Vδ genes are clustered towards the 3′-end of the locus. Based on nucleotide identity shared among the platypus V genes, they can be classified into 17 different Vα families and two different Vδ families, based on the criteria of a V family sharing >80% nucleotide identity (the family and segment numbers are annotated in fig. 3). This is a typical level of complexity for mammalian Vα and Vδ genes.55Giudicelli et al. 2005; Parra et al. 2008
Also present were two Dδ (orange) and seven Jδ (green) gene segments oriented upstream of the Cδ. All gene segments were flanked by canonical recombination signal sequences (RSS), which are the recognition substrate of the RAG recombinase. The D segments were asymmetrically flanked by an RSS containing at 12 bp spacer on the 5′-side and 23 bp spacer on the 3′-side, as has been shown previously for T-cell receptor D gene segments in other species.56Carroll et al. 1993; Parra et al. 2007, 2010 In summary, the overall content and organization of the platypus TCRα/δ locus appeared fairly generic, with one exception.
This atypical feature of the platypus locus is an additional V gene that shares greater identity to antibody VH genes than to T-cell receptor V genes. Among V genes, this segment is the closest to the D and J genes (see the yellow segment in fig. 3). We tentatively designated it as VHδ.
VHδ Phylogenetics
VHδ genes are, by definition, V genes that are indistinguishable from immunoglobulin heavy V (Ig VH) genes, but used in encoding T-cell receptor δ chains. Recall from the introduction[15]Yes, you are allowed to make links between the sections of your paper like this! that they have previously been found only in the genomes of birds and frogs.57Parra et al. 2008, 2010, 2012
To put the platypus VHδ gene in context, let us examine the phylogeny of VH genes. In mammals and other tetrapods, VH genes have been shown to cluster into three ancient clans (shown in fig. 4). Individual species differ in the presence of one or more of these clans in their germ-line immunoglobulin heavy locus.58Tutter and Riblet 1989; Ota and Nei 1994 For example, humans, mice, echidnas, and frogs have VH genes from all three clans,59Schwager et al. 1989; Ota and Nei 1994; Belov and Hellman 2003 whereas rabbits, opossums, and chickens have only a single clan.60McCormack et al. 1991; Butler 1997; Johansson et al. 2002; Baker et al. 2005
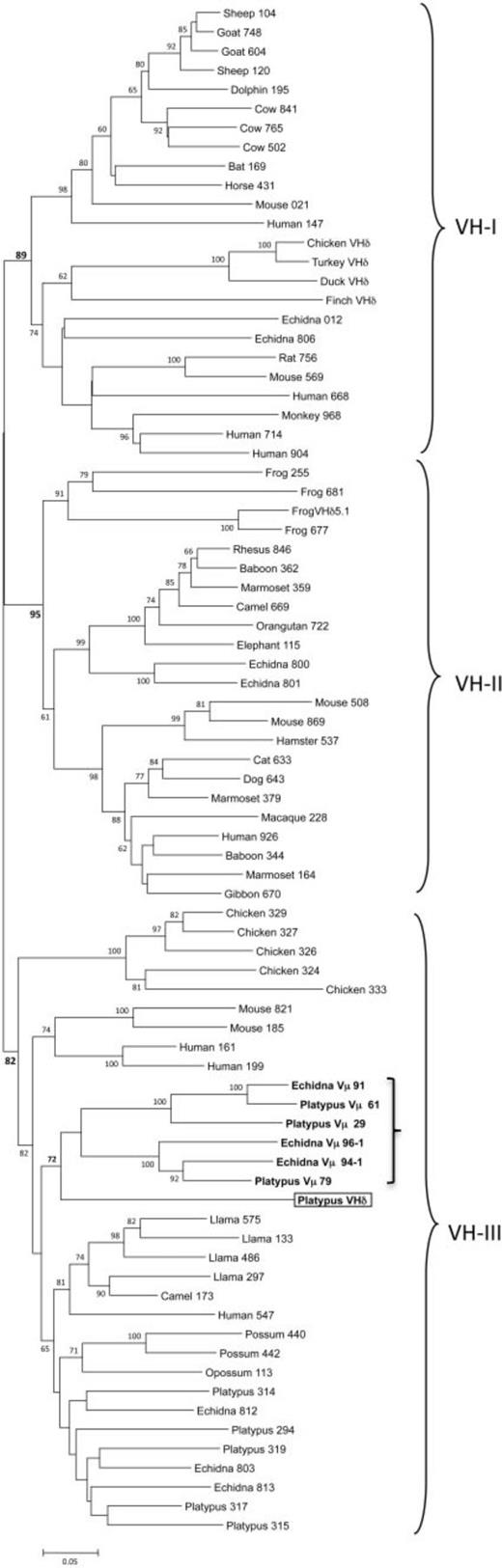
Our phylogenetic analyses showed that the platypus VHδ was most related to the platypus Vµ genes found at the TCRµ locus (see the boxed and bolded parts of fig. 4). Platypus VHδ, however, shares only 51–61% nucleotide identity (average 56.6%) with the platypus Vµ genes. Both the platypus Vµ and VHδ clustered within clan III.61Wang et al. 2011 This is noteworthy since VH genes in the platypus IgH locus are also from clan III and, in general, clan III is the most ubiquitous and conserved lineage of VH.62Johansson et al. 2002; Tutter and Riblet 1989 Although clearly related to platypus VH, the VHδ gene shares only 34–65% nucleotide identity (average 56.9%) with the “authentic”[16]I’m not sure about this but the original phrase was bona fide, which I had to look up. Maybe “authentic” between quotes isn’t the best translation, but a translation is better … Continue reading VH used in antibody heavy chains in this species.
Results of the Confirmation of VHδ Expression
It was necessary to rule out that the VHδ gene present in the platypus TCRα/δ locus was not an artifact of the genome assembly process. This is why we performed a “wet lab” verification step on cDNA synthesized from the splenic RNA of two platypuses, one from New South Wales and one from Tasmania (see Materials and Methods). We performed RT-PCR with primers that were specific for VHδ and Cδ. We were successful in amplifying the PCR products of the NSW specimen, but not for the Tasmanian one.
One piece of supporting evidence for the expression of VHδ would be the demonstration that it is recombined to downstream Dδ and Jδ segments, and expressed with Cδ in complete T-cell receptor δ transcripts. This is what we found from the twenty sequenced clones we obtained from PCR in the New South Wales platypus. Each clone contained a unique nucleotide sequence that comprised the VHδ gene recombined to the Dδ and Jδ gene segments (see fig. 4A). Of these 20, 11 had unique V, D, and J combinations that would therefore encode 11 different complementarity-determining regions-3 (CDR3; see fig. 4B). More then half of these (8 out of 11) contained evidence of using both D genes, giving a VDDJ pattern. This is a common feature of δ V domains where multiple D genes can be incorporated into the recombination due to the presence of asymmetrical RSS.63Carroll et al. 1993

The region corresponding to the junctions between the V, D, and J segments contained an additional sequence that could not be accounted for by the germ-line gene segments (fig. 4B). There are two possible sources of such a sequence. One is palindromic nucleotides that are created during V(D)J recombination when the RAG generates hairpin structures that are resolved asymmetrically during the re-ligation process.64Lewis 1994 The second is non-templated nucleotides that can be added by the enzyme terminal deoxynucleotidyl transferase (TdT) during the V(D)J recombination process.
An unusual feature of the platypus VHδ is the presence of a second cysteine encoded near the 3′-end of the gene, directly next to the cysteine predicted to form the intra-domain disulfide bond in immunoglobulin domains (fig. 4A). Additional cysteines in the complementarity-determining region 3 of VH domains have been thought to provide stability to unusually long CDR3 loops, as has been described for cattle and the platypus previously.65Johansson et al. 2002 The CDR3 of T-cell receptor δ using VHδ are only slightly longer than conventional δ chains (ranging 10–20 residues).66Rock et al. 1994; Wang et al. 2011 Furthermore, the stabilization of CDR3 generally involves multiple pairs of cysteines, which were not present in the platypus VHδ clones (fig. 4A).
The Tasmanian specimen
The above concerns the animal collected from New South Wales. With the Tasmanian specimen, we were unable to amplify T-cell receptor δ transcripts containing VHδ from its splenic cDNA. We did, however, successfully isolate transcripts containing conventional Vα/δ segments, which provides a positive control.
It is possible that Tasmanian platypuses, which have been separated from the mainland population at least 14,000 years ago, either have a divergent VHδ or have deleted this single V gene altogether.67Lambeck and Chappell 2001
Sequence variation in VHδ
Although there is only a single VHδ in the current platypus genome assembly, there was sequence variation in the region corresponding to FR1 through FR3 of the V domains (see fig. 4A; the sequence data are not shown here, but are available in GenBank). We have three potential explanations for this variation:
- Two alleles of a single VHδ gene
- Somatic mutation of expressed VHδ genes
- Allelic variation in gene copy number
The two-allele explanation makes sense given that the RNA used in this experiment is from a wild-caught individual from the same population that was used to generate the whole-genome sequence, which was found to contain substantial heterozygosity.68Warren et al. 2008 However, the variation was too large to be fully explained by this.
The second possibility, somatic mutation (i.e. mutation not occurring in germ cells), is considered controversial for T-cell receptor chains. Nonetheless, it has been invoked in sharks and postulated in salmonids to explain the variation that exceeds the apparent gene copy number in these vertebrates.69Yazawa et al. 2008; Chen et al. 2009 Therefore, it seems possible[17]I kind of like the original phrasing “it does not seem to be out of the realm of possibility” but that could be easily simplified, so I did. that somatic mutation is occurring in platypus VHδ. One piece of evidence in favor of this is that the mutations appear to be localized to the V region with no variation in the C region (fig. 4A). This may be due to the relatedness between VHδ and immunoglobulin VH genes where somatic hyper-mutation is well documented. Somatic mutation in immunoglobulin VH contributes to overall affinity maturation in secondary antibody responses.70Wysocki et al. 1986 However, this means that the evidence is mixed: the pattern of mutation seen in the platypus is found in the complementarity-determining region 3, which would be indicative of selection for affinity maturation, but was also found in the framework regions, which does not indicate this. As further evidence against the somatic mutation explanation, there is no evidence of somatic mutation in the V regions of birds, which also have only a single VHδ.71Parra et al. 2012 The contribution of mutation to the platypus TCRδ repertoire, if it is occurring, remains to be determined.
Alternatively, the sequence polymorphism may be due to VHδ gene copy number variation between individual TCRα/δ alleles.
Irrespective of the number of VHδ genes in the platypus TCRα/δ locus, the results clearly support T-cell receptor δ transcripts containing VHδ recombined to Dδ and Jδ gene segments in the TCRα/δ locus (fig. 4). A VHδ gene or genes in the platypus TCRα/δ locus in the genome assembly, therefore, does not appear to be an assembly artifact. Rather, it is present and functional, and contributes to the expressed T-cell receptor δ chain repertoire. The possibility that some platypus TCRα/δ loci contain more than a single VHδ does not alter the principal conclusions of this study.
Discussion of our Model of the Evolution of TCRα/δ and TCRµ
The results above make up the evidence that allowed us to construct the model shown after the introduction section (see fig. 2). Here we discuss various considerations about the model.
Previous hypothesis of the origin of TCRµ in mammals
Our previous hypothesis72Parra et al. 2008 about the origin of T-cell receptor µ (TCRµ) in mammals involved the recombination between an ancestral TCRα/δ locus and an immunoglobulin heavy (IgH) locus. The IgH locus would have contributed the V gene segments at the 5′-end, while the T-cell receptor δ would have contributed the D, J, and C genes at the 3′-end of the locus.
The difficulty with this hypothesis was the clear stability of the genome region surrounding the TCRα/δ locus. In other words, the chromosomal region containing the TCRα/δ locus appears to have remained relatively undisrupted for at least the past 360 million years.73Parra et al. 2008, 2010, 2012
VHδ in different vertebrate lineages
An alternative model for the origins of TCRµ emerged from the discovery, in amphibians and birds, of VHδ genes inserted into the TCRα/δ locus. This model involves the insertion of VH (fig. 2B) followed by the duplication and translocation of T-cell receptor genes (fig. 2C-E).
The insertion in the TCRα/δ locus seems to occur without disrupting the local syntenic region, as we know from zebra finches and frogs. In frogs, the IgH and TCRα/δ loci are tightly linked, which may have facilitated the translocation of VH genes into the TCRα/δ locus.74Parra et al. 2010
But close linkage is not a requirement. The genomes of birds and platypuses do not show such linkage, and the translocation of VH genes to the TCRα/δ locus appears to have occurred independently from frogs in these two lineages. We know this from the lack of similarity and relatedness between the VHδ genes of frogs, birds, and monotremes.75Parra et al. 2012 As can be seen in the phylogenetic tree of fig. 4, they appear derived each from different, ancient VH clans:
- Clan I for birds
- Clan II for frogs
- Clan III for platypuses
Therefore, we suggest that the transfer of VHδ occurred independently in the different lineages. Another possibility is that transfers of VHδ may have occurred frequently and repeatedly in the past. Gene replacement may be the best explanation for the current content of these genes in the different tetrapod lineages.
The new evidence of platypus VHδ from this study allows us to update the model.
Updating the model for mammalian TCRµ
Let us contrast the evidence from marsupials with the evidence we have gathered from the platypus. In marsupials, there is no VHδ; the Vµ genes are highly divergent; and at least in the opossum, there is no conserved synteny with genes linked to TCRµ. These facts provide little insight into the origins of T-cell receptor µ and its relationship to other T-cell receptor chains like δ or the conventional ones.76Parra et al. 2008
In the platypus genome, however, we notice a striking similarity between VH, VHδ, and Vµ. These genes are all in clan III. In particular, the close relationship between the platypus VHδ and Vµ genes lends greater support for the model presented in fig. 2E, with TCRµ having been derived from TCRδ genes.
The similarity that we found here between the platypus VHδ and V genes in the TCRµ locus is, so far, the clearest evolutionary association between the µ and δ loci in one species.
Evolution of chains with three extracellular domains
TCRµ is an example of a T-cell receptor form with three extracellular domains (refer back to fig. 1). These forms have evolved at least twice in vertebrates. The first was in the ancestors of the cartilaginous fish in the form of NAR-TCR.77Criscitiello et al. 2006 The second was in the mammals as TCRµ.78Parra et al. 2007
As we discussed in the introduction, NAR-TCR uses an N-terminal V domain that is related to the V domains found in IgNAR antibodies, which are unique to cartilaginous fish,79Greenberg et al. 1995; Criscitiello et al. 2006 and not closely related to antibody VH domains. Therefore, it appears that NAR-TCR and TCRµ are more likely the result of convergent evolution rather than being related by direct descent.80Parra et al. 2007; Wang et al. 2011
Evolution of chains with antibody-like V domains
T-cell receptor chains that use antibody-like V domains, such as TCRδ using VHδ, NAR-TCR, or TCRµ (i.e. the receptors with yellow ovals in fig. 1) are widely distributed in vertebrates. Only the bony fish and placental mammals lack them.
In addition to NAR-TCR, some shark species appear to generate T-cell receptor chains using antibody V genes. This occurs via trans-locus V(D)J recombination between immunoglobulin IgM and IgW heavy chain V genes and TCRδ and TCRα D and J genes.81Criscitiello et al. 2010 This may be possible, in part, due to the multiple clusters of immunoglobulin genes found in the cartilaginous fish. It also illustrates that there have been independent solutions to generating T-cell receptor chains with antibody V domains in different vertebrate lineages.
In the tetrapods, the VH genes were trans-located into the T-cell receptor loci where they became part of the germ-line repertoire. By comparison, in cartilaginous fish, something equivalent may occur somatically during V(D)J recombination in developing T cells. Either mechanism suggests there has been selection for having T-cell receptors using antibody V genes over much of vertebrate evolutionary history.
What function do the antibody V chains serve? The current working hypothesis is that they are able to bind native antigen directly. This is consistent with a selective pressure for T-cell receptor chains that may bind or recognize antigen in ways similar to antibodies in many different lineages of vertebrates.
In the case of NAR-TCR and TCRµ, the N-terminal V domain (the “third” one) is likely to be unpaired and bind antigen as a single domain (see fig. 1), as has been described for IgNAR and some IgG antibodies in camels (recently reviewed in Flajnik et al. 2011). This model of antigen binding is consistent with the evidence that the N-terminal V domains in TCRµ are somatically diverse, while the second, supporting V domains have limited diversity and presumably perform a structural role rather than one of antigen recognition.82Parra et al. 2007; Wang et al. 2011
There is no evidence of double V domains in TCRδ chains using VHδ in frogs, birds, or platypus (rightmost part of fig. 1).83Parra et al. 2010, 2012 Rather, the complex containing VHδ would likely be structured similar to a conventional γδ receptors with a single V domain on each chain. It is possible that such receptors also bind antigen directly, but this remains to be determined.
A compelling model for the evolution of the immunoglobulin and T-cell receptor loci has been one of internal duplication, divergence and deletion. This is the so-called birth-and-death model of evolution of immune genes and was promoted by Nei and colleagues.84Ota and Nei 1994; Nei et al. 1997 Our results do not contradict that the birth-and-death mode of gene evolution has played a significant role in shaping these complex loci. However, our results do support the role of horizontal transfer of gene segments between the loci that had not been previously appreciated. With this mechanism, T cells may have been able to acquire the ability to recognize native, rather than processed antigen, much like B cells.
Notes
| ↑1 | This is an example comment. |
|---|---|
| ↑2 | Major changes to the abstract: I split it in four paragraphs with section titles (this is common in some journals; it should be common in most journals). I also added a section at the beginning to state the main point of the paper. Scientists have the bad habit of starting with background information before we even know why we’re supposed to care. This fixes that.
The abstract is longer now, but not terribly so (286 vs. 267 words), so I think it’s fine; it could be shortened some more, but that would be a question of picking what to remove, which I’m less confident to do as I’m not the author. Note that I rewrote the abstract after rewriting the rest. |
| ↑3 | Since this abbreviation comes up a lot, I put it first, with its meaning in parentheses. |
| ↑4 | I didn’t change much in the figure’s caption, but it seemed pretty trivial to add color to the text to facilitate looking up what the colors mean. |
| ↑5 | This is an example of two sentences taken verbatim from the original paper. Not all of it was poorly written! |
| ↑6 | It took me forever to rewrite this part. The original sentence was, “Diversity in antibodies produced by B cells is also generated by RAG-mediated V(D)J recombination and the TCR and Ig genes clearly share a common origin in the jawed-vertebrates.” Soooo many things wrong here.
First, the weirdly formatted term “V(D)J” was not defined anywhere. I assume it means “V, J, and optionally D,” but it’s not as obvious as the authors seem to think. Second, why are we talking about B cells? They don’t come up anywhere else except in the very last sentence of the paper. We’ve been talking about T cells; if you’re going to switch to a different but similarly named type of cell, then you should tell the reader explicitly. Third, this is two different ideas linked together with an “and”. I have no clue why it was written as a single sentence, except maybe for the bad reason of having the citations refer to both ideas. They’re so different that it made sense to split them into not only distinct sentences or paragraphs, but actual sections! |
| ↑7 | The original combined this sentence and the next, even though they’re about quite distinct ideas: the name of the genes, and the species where they’re found. |
| ↑8 | Why not indicate the part of the figure that is relevant? Whenever you can, provide reader guidance! |
| ↑9 | The authors like to use “duckbill platypus,” but there’s only one species of platypus, so I took that word out. |
| ↑10 | Another horrible sentence from the original, recorded for posterity: “Trans-locus V(D)J recombination of V genes from other Ig and TCR loci with TCRµ genes has not been found.” That distance between the subject (recombination) and the verb (has). Ugh. |
| ↑11 | The abbreviation IgSF was used in the paper, with no explanation. I assume the people who would read this paper tend to know what that means, but still. |
| ↑12 | Major change from me here. This section was moved here from the discussion, because it is the core and most interesting part of the paper. It is now its own first-level section alongside Introduction, Materials and Methods, etc.
I also simplified the contents. The six stages used to be identified with letters A-F in the figure, and 1-6 in the text. I changed that to use letters everywhere. I removed most of the figure’s caption since it repeats the text. There was an inconsistency in calling the same thing the Dδ–Jδ–Cδ cluster in the figure and D–J–Cδ cluster in the text. I fixed that. I also color-coded the elements in the text according to the figure. One thing I didn’t like about the original figure is that the six stages aren’t sequential. The figure presented steps A to F as if they followed one another, but steps C, D and E-F describe the evolution in different animal lineages. So I reorganized the contents and added some arrows for clarification. It also seems that the steps 5-6 in the text and E-F in the figure didn’t quite match, with some parts illustrated in step F being explained in step 5; I edited the text so that they do match. I think the figure could be improved much more, notably by splitting the complex F stage in multiple steps, but I don’t want to change it too much. |
| ↑13 | This new paragraph is important! It gives context to the experiments below and it guides the reader for the entire section. Also notice this is a case of an enumeration without point form. I like point form, but it must not be overused. |
| ↑14 | In the original paper, this section comes after the Confirmation of Expression section below, but in the results section, the phylogenetic results are discussed first. I don’t know if there was a reason for this (maybe they performed the phylogenetic analysis later) but it seems better to keep the same order in both sections, which is why I’m placing this part here. |
| ↑15 | Yes, you are allowed to make links between the sections of your paper like this! |
| ↑16 | I’m not sure about this but the original phrase was bona fide, which I had to look up. Maybe “authentic” between quotes isn’t the best translation, but a translation is better than a Latin phrase that many people will not get. |
| ↑17 | I kind of like the original phrasing “it does not seem to be out of the realm of possibility” but that could be easily simplified, so I did. |





